Bartoli indole synthesis
Bartoli吲哚合成法
重要性
[开创性文献1-3;综述4-7;改进与优化8-11]
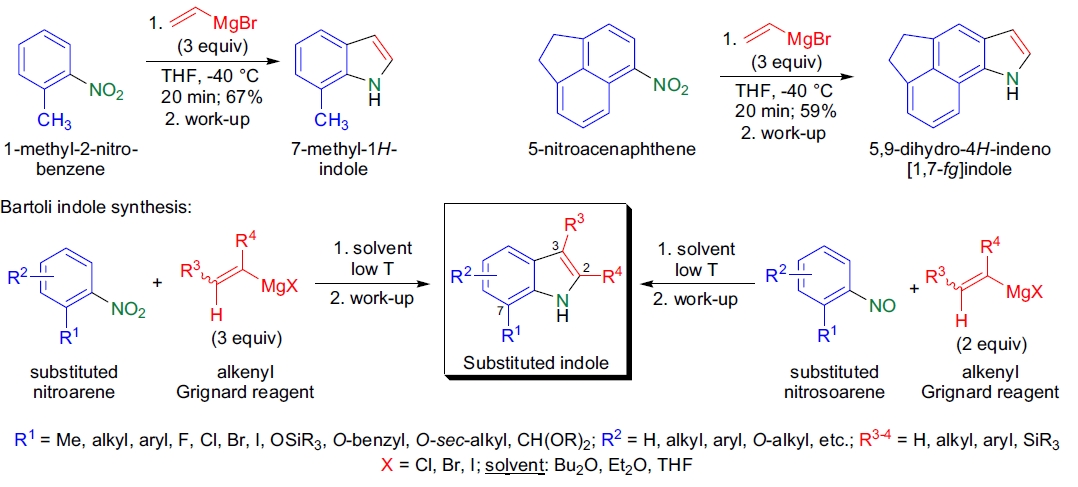
Bartoli et al. 在1989年描述了一种反应,通过在低温下将取代的硝基芳烃与过量的乙烯基Grignard试剂反应,经水工作后生成取代的吲哚2。该反应通常使用邻位取代的硝基芳烃,并通过乙烯基Grignard试剂的反应生成7位取代的吲哚,称为Bartoli吲哚合成。邻位没有取代基的硝基芳烃反应产率低甚至没有产物,而邻位取代基越大,产率越高。此外,除乙烯基Grignard外,取代的烯基Grignard也可用于生成C2或C3取代的吲哚。当底物是硝基芳烃时,需要三当量的Grignard试剂,而对于亚硝基芳烃,仅需要两当量。
机理12,7
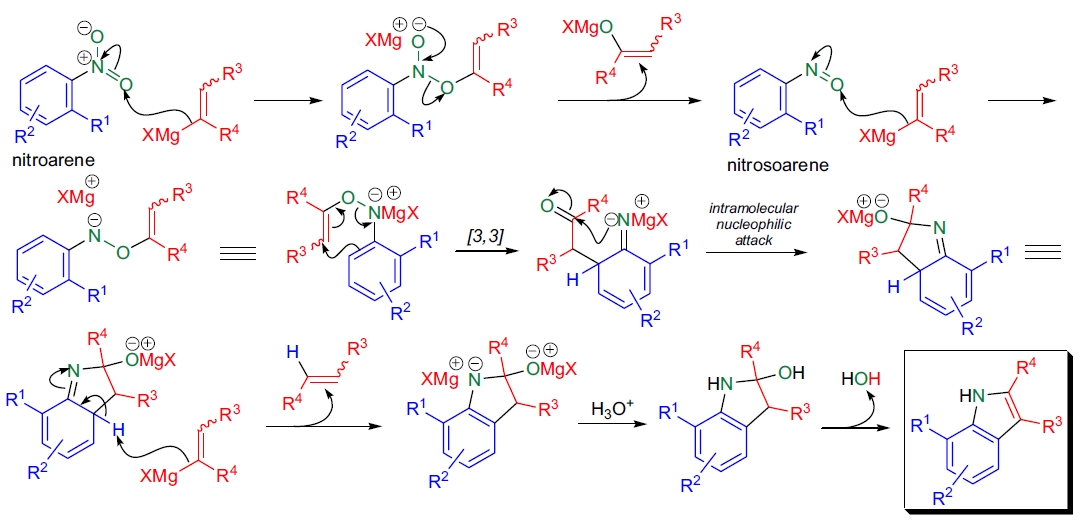
该反应的详细机理尚未完全清楚,但Bartoli和同事成功阐明了主要步骤:Grignard试剂首先进攻硝基氧原子,形成O-烯基化产物,迅速分解生成亚硝基芳烃。亚硝基芳烃被第二当量Grignard试剂进攻,生成O-烯基羟胺衍生物,该羟胺衍生物发生[3,3]-σ迁移,迁移产物经分子内亲核进攻生成环化产物,第三当量Grignard试剂去除环中质子,最终酸性工作后生成吲哚。
合成应用
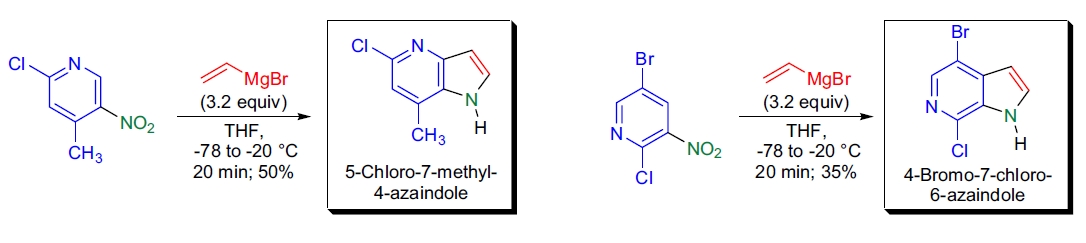
1. 4-和6-氮吲哚的合成: T. Wang实验室基于Bartoli吲哚合成开发了一种制备4-和6-氮吲哚的方法13。该方法使用乙烯基Grignard试剂处理取代硝基吡啶,发现邻位取代基越大,产率越高。
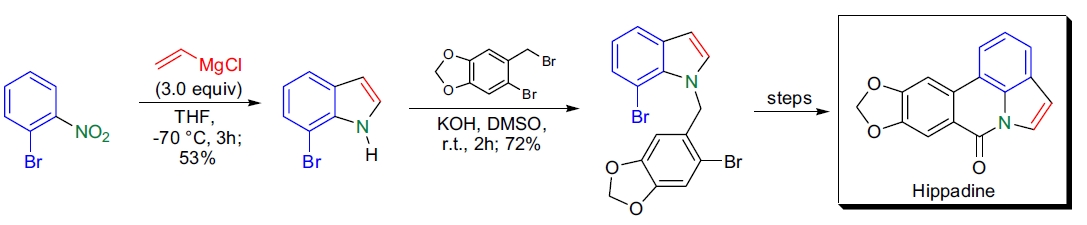
2. Hippadine的短合成: D.C. Harrowven等通过Bartoli吲哚合成制备了7-溴吲哚,并用于Ziegler改进的分子内Ullmann偶联制备pyrrolophenanthridone生物碱hippadine14。
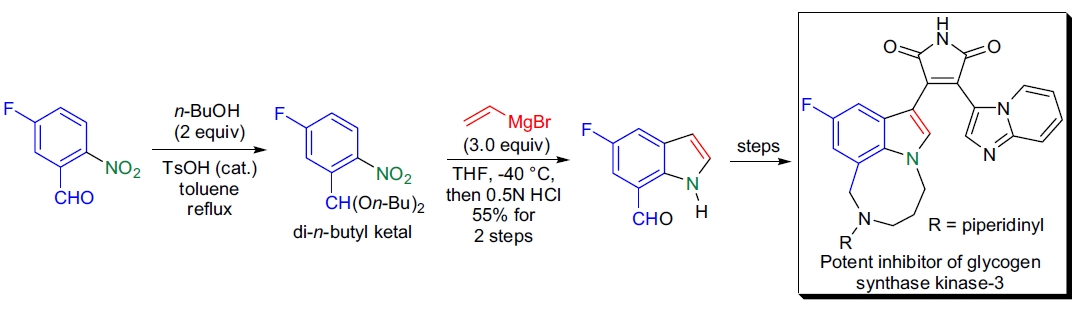
3. GSK3抑制剂的合成: T.A. Engler等制备了选择性糖原合酶激酶-3(GSK3)抑制剂,通过Bartoli吲哚合成获得5-氟-7-甲酰吲哚15。采用两步法保护5-氟-2-硝基苯甲醛的甲酰基,然后使用Grignard试剂反应,最后脱去保护基。
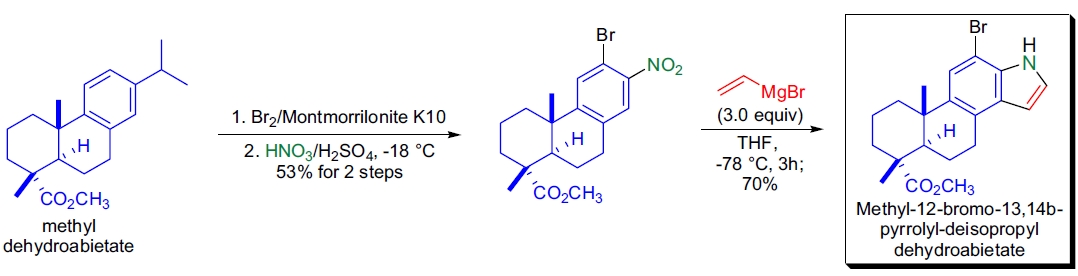
4. 抗病毒化合物的合成: B. Gigante等从脱氢松香酸出发,通过Bartoli吲哚合成制备多种杂环化合物,并评估其抗病毒活性16。脱氢松香酸首先酯化、溴化和硝基脱异丙基化处理,然后与乙烯基Grignard试剂反应得到目标化合物。