产物官能团类型
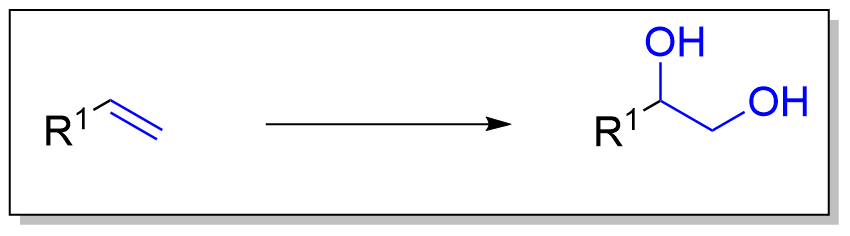
双羟化简介
双羟化是一种将烯烃转化为邻二醇的过程。虽然有许多方法可以实现这种氧化,但最常见和直接的方法是使用高氧化态的过渡金属(通常是锇或锰)。金属通常用作催化剂,同时需要某些化学计量的氧化剂。[1] 此外,还开发并应用了其他过渡金属和非过渡金属的方法来催化该反应。
四氧化锇(OsO4)因其在生成顺式二醇方面的可靠性和高效性而成为烯烃二羟基化中常用的氧化剂。由于其价格昂贵且有毒,仅使用催化量的OsO4,并结合化学计量的氧化剂。[2][3] 米拉斯羟基化(Milas hydroxylation)、Upjohn二羟基化以及Sharpless不对称二羟基化反应均以锇作为催化剂,并结合不同的次级氧化剂。
1930年提出的米拉斯二羟基化(Milas dihydroxylation)使用过氧化氢作为化学计量氧化剂。[4] 尽管该方法可以生成二醇,但由于过氧化反应生成二羰基化合物,导致难以分离得到邻二醇。[4] 因此,米拉斯方案被Upjohn和Sharpless不对称二羟基化所取代。
Upjohn二羟基化于1973年提出,使用OsO4作为二羟基化过程中活性催化剂,同时采用N-甲基吗啉N-氧化物(NMO)作为化学计量氧化剂以再生锇催化剂,从而仅需使用催化量的锇。[2][5] Upjohn方法对邻二醇的转化率较高,并能适用于多种底物。然而,该方法无法对四取代烯烃进行二羟基化。[2] Upjohn条件可用于从烯丙醇合成反式二醇,正如Kishi及其同事所示。[6]
参考文献:
1. Carey, Francis A.; Sundberg, Richard J. Advanced Organic Chemistry Part B: Reactions and Synthesis (5th ed.). Springer.
2. Schroder, M. (1980). "Osmium tetraoxide cis hydroxylation of unsaturated substrates". Chem. Rev. 80 (2): 187–213. doi:10.1021/cr60324a003
3. Kolbe, H.C.; VanNieuwanhze, M.S.; Sharpless, K.B. (1994). "Catalytic Asymmetric Dihydroxylation". Chem. Rev. 94 (8): 2483–2547. doi:10.1021/cr00032a009
4. Milas, N.A.; Sussman, S. (1936). "The Hydroxylation of the Double Bond 1". J. Am. Chem. Soc. 58 (7): 1302–4. doi:10.1021/ja01298a065
5. Dupau, P.; Epple, R.; Thomas, A.A.; Fokin, V.V.; Sharpless, K.B. (2002). "Osmium-Catalyzed Dihydroxylation of Olefins in Acidic Media: Old Process, New Tricks". Adv. Synth. Catal. 344 (3–4): 421–33. doi:10.1002/1615-4169(200206)344:3/4<421::AID-ADSC421>3.0.CO;2-F
6. Cha, J.K.; Christ, W.J.; Kishi, Y. (1983). "1983". Tetrahedron Lett. 24: 3943–6. doi:10.1016/s0040-4039(00)88231-3
1. OsO4 + NMO
四氧化锇(OsO4)与N-甲基吗啉N-氧化物(NMO)作为助氧化剂一起使用时(Upjohn双羟化反应),仅需催化量的有毒OsO4。常见的溶剂组合包括丙酮/水和叔丁醇/水。[1][2]
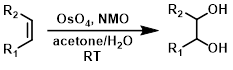
案例
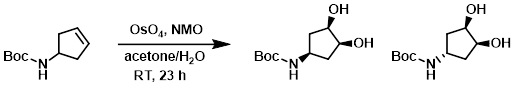
To a solution of the SM (1.94 g, 10.58 mmol) in a mixture of acetone (63 mL) and H2O (7.8 mL) at RT was added NMO (2.48 g, 21.16 mmol). After 2 min, OsO4 (4% in H2O, 3.37 mL) was added and the rxn mixture was stirred at RT for 23 h. The mixture was quenched with aq 0.2M Na2S2O3 (25 mL) and extracted with DCM (2 x 50 mL). The combined organics were washed with aq 0.2M Na2S2O3 (10 mL), dried (MgSO4), and concentrated in vacuo. The crude material was purified by flash chromatography (17-100% EtOAc/heptane) to provide the product as a yellow solid. [1.31 g, 57%]
Patent Reference: WO2016001341, page 126
2.Sharpless不对称双羟化
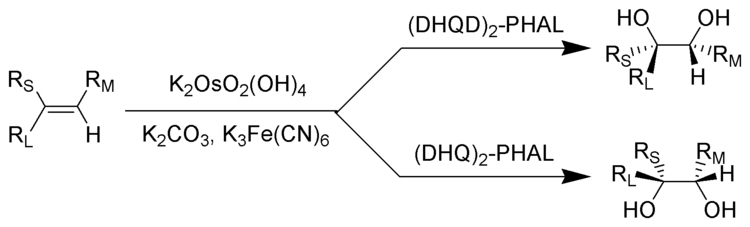
案例

A 3-L flask with a mechanical stirrer, thermometer and an inlet port open to the atmosphere is charged with 375 mL of water. Stirring is started and the following reagents are added in the order indicated through a powder funnel: potassium ferricyanide (247 g, 0.75 mol, 3 equiv), anhydrous potassium carbonate (104 g, 0.75 mol, 3 equiv), methanesulfonamide (23.8 g, 0.25 mol, 1 equiv), potassium osmate dihydrate (46.1 mg, 0.125 mmol, 0.05 mol %), (DHQD)2PHAL [1,4-bis(9-O-dihydroquinidinyl)phthalazine, 486.9 mg, 0.625 mmol, 0.25 mol %], 1-phenylcyclohexene (1, 39.55 g, 0.25 mol) and tert-butyl alcohol (250 mL) (Notes 1 and 2). The slurry is stirred vigorously for 2 days at a rate of 500 rpm. During this time, the product crystallizes in the top organic phase, beginning after 4 hr. Also, the appearance of the slurry gradually changes from a mixture containing red granules (ferricyanide) to yellow flakes, which are presumably a salt of iron(II).
After the reaction is complete the mixture is treated with ethyl acetate (250 mL) with stirring to dissolve the product. The resulting mixture is filtered through a 500-mL medium-fritted glass funnel and the flask and filter cake are washed with ethyl acetate (3 × 50 mL). The filtrate is transferred to a 2-L separatory funnel and the brown-colored aqueous phase is separated. The organic phase is washed with 2 M potassium hydroxide (KOH, 2 × 50 mL) with vigorous shaking to remove methanesulfonamide , then dried over magnesium sulfate (MgSO4, 12.5 g). The solid is filtered, the cake is washed with ethyl acetate (2 × 37 mL) and the filtrate is evaporated, to afford a white solid. After the crude product is dried under reduced pressure overnight, it weighs 47.44 g (99%) (Notes 3 and 4).