Still-Gennari 改进的 HWE 烯化反应
重要性
[开创性文献1;综述2,3;改进与优化4-10;理论研究11]
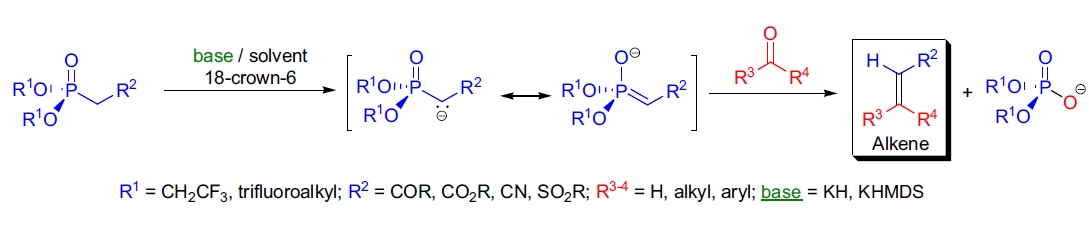
Horner-Wadsworth-Emmons (HWE) 烯化反应与 wittig反应 反应是制备 (E)-烯烃的两种主要方法。HWE 烯化反应生成 (E)-α,β-不饱和酮和酯,而trans选择性的 Wittig 反应则生成简单的、非共轭的 (E)-烯烃。
1983年,W.C. Still 和 C. Gennari 首次通过对 HWE 烯化中的膦酸酯试剂进行改进,实现了从醛出发制备 (Z)-烯烃的通用方法。该方法利用含三氟乙氧基的膦酸酯与醛在强碱存在下偶联生成 (Z)-α,β-不饱和酮和酯,被称为 Still-Gennari 改进的 HWE 烯化反应。
反应的主要特点包括:
- 必要的双三氟乙氧基膦酸酯可通过商品化的三烷基膦酸酯和三氟乙醇轻松制备;
- (Z)-选择性不仅适用于 1,2-二取代烯烃,也适用于三取代烯烃;
- 膦酸酯试剂必须在 α-位具有电子吸引(稳定碳负离子)的基团,否则膦酸酯碳负离子易分解;
- 必须使用易解离的碱,其金属阳离子不可与试剂发生配位(通常通过在反应混合物中加入 18-冠-6 实现);
- 当 R₂=CN 时,(Z)-选择性较高,而 α-氰稳定的常规膦酸酯对 (E)-选择性较差。
机理
[参考文献12]
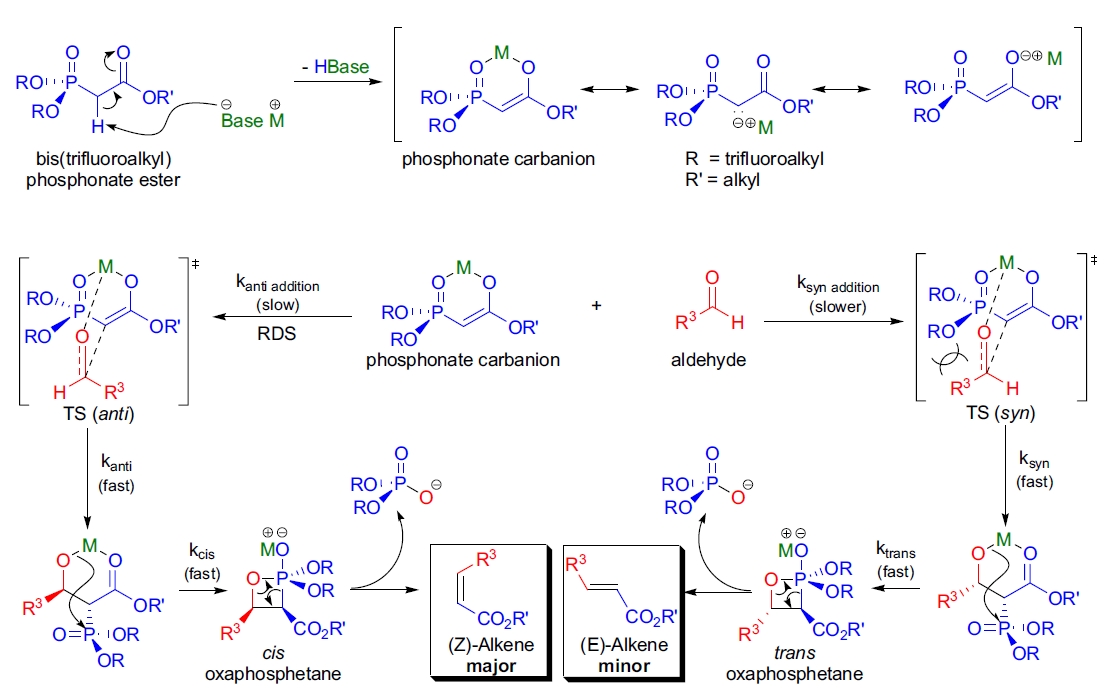
HWE 烯化反应的机理尚未完全阐明。在 Still-Gennari 改进的 HWE 烯化反应中,由于磷上有两个电子吸引的三氟乙氧基,螯合加成物向氧磷杂四元环的重排更容易发生,且消除步骤比初始加成快,这使得反应基本不可逆(不同于常规 HWE 烯化反应)。因此,(Z)-立体异构体的形成占主导地位。
合成应用
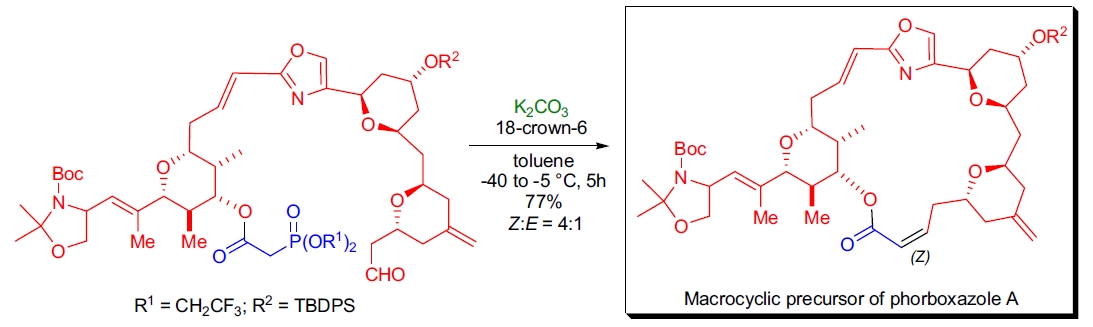
1. Phorboxazole A 的全合成: C.J. Forsyth 团队利用 Still-Gennari 改进的 HWE 烯化反应的分子内版本对复杂双三氟乙氧基膦酸酯醛前体进行大环化反应。该前体溶于甲苯,并在 K₂CO₃ 和 18-冠-6 存在下反应,生成目标 C1-C3 (Z)-丙烯酸酯部分,产率为 77%,(Z:E) 比为 4:113。
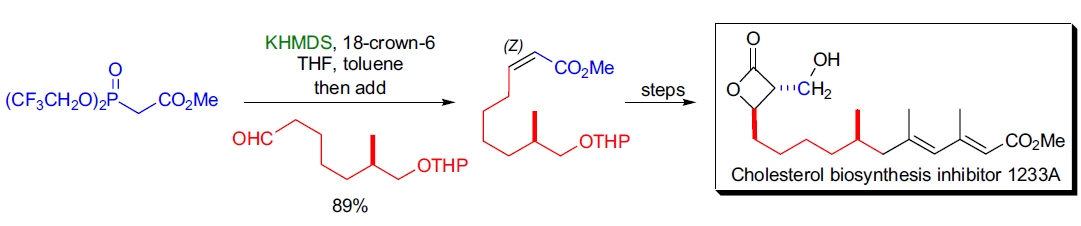
2. β-内酯胆固醇合酶抑制剂 1233A 的全合成: S.V. Ley 团队通过氧化解络合实现了 (π-烯丙基)三羰基铁内酯的关键步骤。目标分子中的 (Z)-烯烃由含双三氟乙氧基膦酸酯和醛的 S-G 改进的 HWE 烯化反应制备,生成目标 α,β-不饱和甲酯,产率优异14。
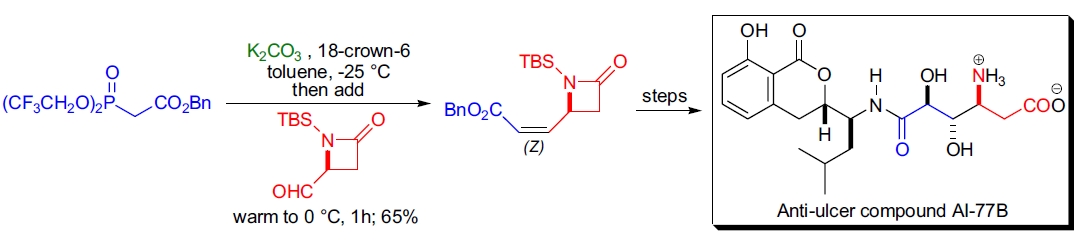
3. 抗溃疡药物 AI-77B 的对映选择性合成: E.J. Thomas 团队通过 Still 改进的 HWE 烯化反应,从 4-甲酰基乙烯酮出发合成了 4-(Z)-烯基氮杂环丁酮的对映选择性合成。用于反应的苯基双三氟乙氧基膦酸酯通过磷酸二氯化物和三氟乙醇制备,并用苄基溴乙酸酯烷基化15。
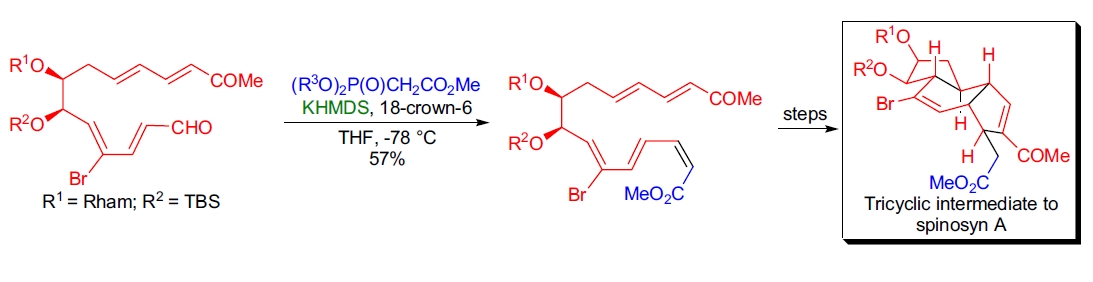
4. Spinosyn A 的全合成: W.R. Roush 团队通过一次性串联分子内 Diels-Alder 反应和分子内扩展 Baylis-Hillman 环化构建了关键三环中间体。环化前体通过 S-G 改进的 HWE 烯化反应制备16。